List of all input keywords¶
'[Trial]' : These options are not tested well
&calculation¶
- theory (character, default='')
- Choice of Calculation theories.Options
dft/ ground state calculation based on DFTdft_md/ adiabatic ab initio MD simulations based on DFTtddft_response/ simulations based on TDDFT for responsetddft_pulse/ simulations based on TDDFT using pulsed lightsingle_scale_maxwell_tddft/ coupled Maxwell and TDDFT single-scale simulationmulti_scale_maxwell_tddft/ coupled Maxwell and TDDFT multi-scale simulationmaxwell/ electromagnetic simulations based on the Maxwell's equationsdft_k_expand/ convert checkpoint data of dft with k-points calculation to that of larger supercell system with gamma-point
- yn_md (character, Default='n')[Trial]
- Available for
theory='dft'(Adiabatic ground-state MD) andtheory='tddft_pulse'(Ehrenfest MD).Molecular dynamics option.Options'y'/ enable'n'/ disable
- yn_opt (character, Default='n')[Trial]
- Available for
theory='dft'.Geometry optimization option.Options'y'/ enable'n'/ disable
&control¶
- sysname (character, Default='default')
- Available for all options of
theory.Name of calculation. This is used for a prefix of output files.
- base_directory (character, Default='./')
- Available for all options of
theory.Name of a default directory, where the basic results will be written down.
- yn_restart (character, Default='n')
- Available for
theory='dft*' or '*tddft*'.Restart option.Options'y'/ enable'n'/ disable
- directory_read_data (character, Default='restart/')
- Directory name for the restart data that is written down in the previous run
- yn_self_checkpoint (character, Default='n')
- If set 'y': When saving intermediate results of the simulation (this call checkpointing), each process write/read a checkpoint data independently.This option helps large-scale simulation to recover from system failure, which reduce restart costs.
- checkpoint_interval (integer, Default=-1)
- Available for
theory='dft*' or '*tddft*'.Interval of time step (or iteration step) of writing down check-point data during the time-propagation or iteration. These are not written down If negative value is set.
- yn_reset_step_restart (character, Default='n')
- Available for
yn_restart='y'with the DFT/TDDFT based options oftheory.In the case of restarting, the initial step of SCF iteration (for DFT) or time step (for TDDFT) are reset to 0 at begining. Then, the memory of the density in the previous SCF iteration steps (in GS) is abondoned.
- read_gs_restart_data (character, Default='all')
- Available for
yn_restart='y'withtheory='dft'.Optionsall/ all of restart data are readall:single/ same asalloption but single file format is read even though the check-point data format is specified byyn_self_checkpoint='y'rho_inout/ only electron densities including memories at previous iteration steps are read (rho_inout.bin file)rho_inout:single/ same asrho_inoutoption but single file format is read even though the check-point data format is specified byyn_self_checkpoint='y'rho/ only the latest electron density is read (user-made data)wfn/ only wavefunctions is readSpecified data which is included in the restart (or checkpoint) directory generated in the previous calculation is used for restarting SCF iteration in DFT. The default option
'all'gives the complete restart. The other options use a part of restart data (other necessary data is generated as done in the initial SCF step)
- write_gs_restart_data (character, Default='all')
- Available for
theory='dft'.Optionsall/ all of restart data are written outrho_inout/ only electron densities including memories at previous iteration steps are written outwfn/ only wavefunctions is written outcheckpoint_only/ the restart data is printed only in the check-point data format (separated data for each process) at the last step (yn_self_checkpoint='y'is required)Specified data is written out in the restart (or checkpoint) directory. The default option
'all'gives the complete set of restart data.
- time_shutdown (real(8), Default=-1d0)[Trial]
- Available for
theory='dft' or '*tddft*'.Timer for automatic shutdown. The unit is second. If negative time is chosen, the automatic shutdown is not performed.
- method_wf_distributor (character, Default='single')
- Available for
theory='dft*' or '*tddft*'.Select a method of save/load the wave function.'single': all wave functions are saved(loaded) to(from) a single file.'slice' : each orbital function is saved(loaded) to(from) a file.'slice' reduces I/O costs, and they can helps flexible large-scale simulation.
- nblock_wf_distribute (integer, Default='16')
- Available for
method_wf_distributor='slice'.'slice' mode savesnblock_wf_distributefiles to one directory.
&units¶
- unit_system (character, Default='au')
- Units of input variables.Options
'au'or'a.u.'/ atomic unit system.'A_eV_fs'/ Angstrom-eV-fs unit system
¶llel¶
- nproc_k/nproc_ob/nproc_rgrid(3) (integer, Default=0)
- Options
nproc_k/ Number of MPI parallelization for orbitals that related to the wavefunction calculation.nproc_ob/ Number of MPI parallelization for orbitals that related to the wavefunction calculation.nproc_rgrid(3)'/ Number of MPI parallelization for each direction in real-space that related to the wavefunction and the electron density calculations.Defaults are0fornproc_k/nproc_oband(0,0,0)fornproc_rgrid. If users use the defaults, automatic proccess assignment is done. Users can also specifynproc_k,nproc_ob, andnproc_rgridmanually. In that case,nproc_kmust be set to1for isolated system calculations.nproc_kandnproc_kmust be set to1fortheory='maxwell'. In addition, followings must be satisfied.nproc_k*nproc_ob*nproc_rgrid(1)*nproc_rgrid(2)*nproc_rgrid(3)= total number of processes.
- yn_ffte (character, Default='n')
- Available for
&system/yn_periodic='y'Method of Fourier transformation.Enable('y')/disable('n').SALMON uses FFT (via FFTE library) to solve poisson equation.When enabling it, followings must be satisfied.mod(num_rgrid(1), nproc_rgrid(2)) == 0mod(num_rgrid(2), nproc_rgrid(2)) == 0mod(num_rgrid(2), nproc_rgrid(3)) == 0mod(num_rgrid(3), nproc_rgrid(3)) == 0
- yn_scalapack (character, Default='n')
- Available for
&calculation/theory='dft' or 'dft_md'SALMON uses ScaLAPACK library to solve eigenvalue problem in subspace diagonalization.When enabling it, you should build SALMON by linking ScaLAPACK library.Options'y'/ enable'n'/ disable
- yn_eigenexa (character, Default='n')
- Available for
&calculation/theory='dft' or 'dft_md'SALMON uses RIKEN R-CCS EigenExa library to solve eigenvalue problem in subspace diagonalization.When enabling it, you should build SALMON by linking ScaLAPACK and EigenExa libraries.Options'y'/ enable'n'/ disable
- yn_diagonalization_red_mem (character, Default='n')
- Available for
¶llel/yn_scalapack='y'or¶llel/yn_eigenexa='y'We use ScaLAPACK/EigenExa libraries by optimized algorithm to reduce memory consumption.Options'y'/ enable'n'/ disable
- process_allocation (character, Default='grid_sequential')
- You can select the process allocation ordering.Options
'grid_sequential'/ real-space grid major ordering.'orbital_sequential'/ orbital-space major ordering.Suggestion:&calculation/theory='dft' or 'dft_md'/ orbital_sequential&calculation/theory='tddft*' or '*maxwell_tddft'/ grid_sequential
&system¶
- yn_periodic (character, Default='n')
- Available for all options of
theory.Option of periodic boundary condition.'y'/ periodic systems (solids)'n'/ isolated systems
- spin (character, Default='unpolarized')
- Available for all options of
theoryexcept fortheory='maxwell'.Variable for classification of spin-unpolarized (closed shell) systems and spin-polarized (open shell) systems.Options'unpolarized'/ spin-unpolarized systems (default)'polarized'/ spin-polarized systems
- al(3) (real(8), Default=0d0)
- Available for all options of
theoryexcept fortheory='maxwell'.Spatial grid box size or lattice constants for cuboid cell (x, y, z). For nonorthogonal cell, see
al_vec1,al_vec2,al_vec3.
- al_vec1(3)/al_vec2(3)/al_vec3(3) (real(8), Default=0d0)
- Available for all options of
theoryexcept fortheory='maxwell'.Primitive lattice vectors for nonorthogonal cell.
- nstate (integer, Default=0)
- Available for the DFT/TDDFT based options of
theory.Number of orbitals/bands.
- nelec (integer, Default=0)
- Available for the DFT/TDDFT based options of
theory.Number of valence electrons.
- nelec_spin(2) (integer, Default=0)
- Available for the DFT/TDDFT based options of
theory.Number of up/down-spin electrons can be specified for each by
nelec_spin(1)/nelec_spin(2). This option is incompatible withnelec(ifnelec_spinis specified,nelecis ignored.)
- temperature (real(8), Default=-1d0)
- Available for DFT-based options of
theoryTemperature of electrons. The value must be given by the unit of energy as specified in&units/unit_system.The kelvin unit can be used by the keywordtemperature_k(see next).temperature < 0/ the occupation numbers are fixed bynelec(for bandgap system).temperature = 0/ redistribution of the occupation numbers by the step function.temperature > 0/ redistribution of the occupation numbers by the Fermi-Dirac distribution function.
- temperature_k (real(8), Default=-1d0)[Trial]
- Available for DFT-based options of
theoryThe same as
temperaturebut in kelvin.
- nelem (integer, Default=0)
- Available for the DFT/TDDFT based options of
theory.Number of used atomic elements in the system.
- natom (integer, Default=0)
- Available for the DFT/TDDFT based options of
theory.Number of atoms in the system.
- file_atom_red_coor (character, Default='none')[Trial]
- Available for the DFT/TDDFT based options of
theory.File name for atomic positions given in reduced coordinates. This option is incompatible with
&system/file_atom_coor,&atomic_coor, and&atomic_red_coor.
- file_atom_coor (character, Default='none')[Trial]
- Available for the DFT/TDDFT based options of
theory.File name for atomic Cartesian coordinates (The unit is specified by
&units/unit_system). This option is incompatible with&system/file_atom_coor,&atomic_coor, and&atomic_red_coor. (XXX why this keyword is not in &atomic_coor ?? XXX)
&atomic_red_coor¶
Atomic coordinates in reduced coordinates as following format:
Here, the information of atoms is ordered in row.
For example, the first row is for the first atom.
The number of rows must be equal to &system/natom.
The first coloum can be any caracters and does not affect calculations.
The second, third and fourth columns are reduced coordinates for
the first, second and third directions, respectively.
The fifth column is a serial number of the atom spieces, which is defined in &pseudo.
This option is incompatible with
&system/file_atom_red_coor, &system/file_atom_coor, and &atomic_coor.
&atomic_coor¶
Cartesian atomic coordinates.
The format is the same as &atomic_red_coor.
The unit can be chosen by &units/unit_length.
This option is incompatible with
&system/file_atom_red_coor, &system/file_atom_coor, and &atomic_red_coor.
&pseudo¶
Input for psudopotentials. Size of array (:) is equal to &system/nelem.
- izatom(:) (integer, Default=-1)
- Available for the DFT/TDDFT based options of
theory.Atomic number.
- file_pseudo(:) (character, Default='none')
- Available for the DFT/TDDFT based options of
theory.File name for pseudopotential.
- lmax_ps(:) (integer, Default=-1)
- Available for the DFT/TDDFT based options of
theory.Maximum angular momentum of pseudopotential projectors. If not given, it is automatically read from the pseudopotential file.
- lloc_ps(:) (integer, Default=-1)
- Available for the DFT/TDDFT based options of
theory.Angular momentum of pseudopotential that will be treated as local.
- yn_psmask(:) (character, Default='n')[Trial]
- Available for the DFT/TDDFT based options of
theory.Fourier filtering for pseudopotentials.Enable(
'y')/disable('n')
- alpha_mask(:) (real(8), Default=0.8d0)[Trial]
- Available for the DFT/TDDFT based options of
theory.Parameter for the Fourier filtering for pseudopotential.
- gamma_mask(:) (real(8), Default=1.8d0)[Trial]
- Available for the DFT/TDDFT based options of
theory.Parameter for the Fourier filtering for pseudopotential.
- eta_mask(:) (real(8), Default=15.0d0)[Trial]
- Available for the DFT/TDDFT based options of
theory.Parameter for the Fourier filtering for pseudopotential.
&functional¶
- xc (character, Default='none')
- Available for the DFT/TDDFT based options of
theory.Exchange-correlation functionals.At present version, the functional 'PZ', 'PZM' and 'TBmBJ' is available for both 0d/3d calculations, and the functionals 'TPSS' and 'VS98' are available for 3d calculations. (XXX need check XXX)Options'PZ': Perdew-Zunger LDA :Phys. Rev. B 23, 5048 (1981).'PZM': Perdew-Zunger LDA with modification to improve sooth connection between high density form and low density one. :J. P. Perdew and Alex Zunger, Phys. Rev. B 23, 5048 (1981).'TBmBJ': Tran-Blaha meta-GGA exchange with Perdew-Wang correlation. :Fabien Tran and Peter Blaha, Phys. Rev. Lett. 102, 226401 (2008). John P. Perdew and Yue Wang, Phys. Rev. B 45, 13244 (1992).'TPSS': Tao, Perdew, Staroverov and Scuseria meta-GGA exchange correlation. :J. Tao, J. P. Perdew, V. N. Staroverov, and G. E. Scuseria, Phys. Rev. Lett. 91, 146401 (2003).'VS98': van Voorhis and Scuseria exchange with Perdew-Wang correlation: T. Van Voorhis and G. E. Scuseria, J. Chem. Phys. 109, 400 (1998).
- cname, xname (character, Default='none')
- Available for
theory='XXX'.XXX
- alibxc, alibx, alibc (character, Default='none')
- Available for the DFT/TDDFT based options of
theory.By specifying
alibxc, the functionals prepared in libxc package are available. They can be set indivisually by specifyingalibxandalibc. To use libxc libraries,--enable-libxcoption must be added in excecuting configure. The available option of the exchange-correlation functionals are listed in the LibXC website. [See http://www.tddft.org/programs/libxc/functionals/]
- cval (real(8), Default=-1d0)
- Available for
xc='TBmBJ'.Mixing parameter in Tran-Blaha meta-GGA exchange potential. If
cvalis set to a minus value, the mixing-parameter computed by the formula in the original paper [Phys. Rev. Lett. 102, 226401 (2008)]. Default is estimated from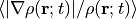 .
.
&rgrid¶
- dl(3) (real(8), Default=0d0)
- Available for the DFT/TDDFT based options of
theory.Spacing of real-space grids. (This cannot be used together with
&rgrid/num_rgrid.)
- num_rgrid(3) (integer, Default=0)
- Available for the DFT/TDDFT based options of
theory.Dividing number of real-space grids for each direction. (This cannot be used together with
&rgrid/dl.)
&kgrid¶
- num_kgrid(3) (integer, Default=1)
- Available for
yn_periodic='y'with the DFT/TDDFT based options oftheory.Number of k-points (grid points of k-vector) for each direction discretizing the Brillouin zone.
- file_kw (character, Default='none')
- Available for
yn_periodic='y'with the DFT/TDDFT based options oftheory.File name for user specified k-points. This file will be read ifnum_kgridis smaller than 1. The k-points are given as following format, for example, :8 #(number of k-points)1 -0.50 -0.50 -0.50 0.1250 #(id, kx, ky, kz, weight)2 -0.50 -0.50 0.00 0.12503 -0.50 0.00 -0.50 0.12504 -0.50 0.00 0.00 0.12505 0.00 -0.50 -0.50 0.12506 0.00 -0.50 0.00 0.12507 0.00 0.00 -0.50 0.12508 0.00 0.00 0.00 0.1250
&tgrid¶
- nt (integer, Default=0)
- Available for 'dft_md' and TDDFT-based options of
theory.Number of total time steps for real-time propagation.
- dt (real(8), Default=0d0)
- Available for 'dft_md' and TDDFT-based options of
theory.Time step size.
- gram_schmidt_interval (integer, Default=-1)
- Available for TDDFT-based options of
theory.Interval of time step for the Gram-Schmidt orthonormalization of the orbital wavefunctions in the time-evolution calculation. If this is set to zero, it is used at the initial step only.
&propagation¶
- n_hamil (integer, Default=4)
- Available for TDDFT-based options of
theory.Order of Taylor expansion of a propagation operator.
- propagator (character, Default=middlepoint')
- Available for TDDFT-based options of
theory.Propagator (time-integrator).Optionsmiddlepoint/ propagator with the Hamiltoinan at midpoint of two-times.aetrs/ time-reversal symmetry propagator.[M.A.L. Marques, A. Castro, G.F. Bertsch, and A. Rubio, Comput. Phys. Commun., 151 60 (2003)].
- yn_predictor_corrector (character(1), Default='n')
- Available for TDDFT-based options of
theory.Switch of the predictor-corrector method of TDDFT.For meta-GGA functionals (xc='tbmbj'or'bj_pw'), the predictor corrector is automatically used even withyn_predictor_corrector='n'.Options'y'/ enable'n'/ disable
- yn_fix_func (character(1), Default='n')[currently not available]
- Available for 'dft_md' and TDDFT-based options of
theory.Option not to update functional (or Hamiltonian) in time-evolution, i.e., keep ground state Hamiltonian. (currently not available)Options'y'/ enable'n'/ disable
&scf¶
- method_init_wf (character, Default='gauss')
- Available for 'dft' and 'dft_md' options of
theory.The generation method of the initial wavefunction (orbital) at the begening of the SCF iteration in DFT calculation.Optionsgauss/ put single gauss function using a random number on each initial orbitalgauss2/ put two gauss functions using a random number on each initial orbitalgauss3/ put three gauss functions using a random number on each initial orbitalgauss4/ put four gauss functions using a random number on each initial orbitalgauss5/ put five gauss functions using a random number on each initial orbitalgauss10/ put ten gauss functions using a random number on each initial orbitalrandom/ give a random number at each real-space grid point on each initial orbital
- iseed_number_change (integer, Default=0)
- Available for 'dft' and 'dft_md' options of
theory.The seed of the random numbers are changed by adding the given number for generating the initial wavefunctions.
- nscf (integer, Default=300)
- Available for 'dft' and 'dft_md' options of
theory.Number of maximum SCF cycle in DFT calculation.
- method_min (character, Default='cg')
- Available for 'dft' and 'dft_md' options of
theory.Method for SCF iterationOptionscg/ Conjugate-Gradient(CG) methoddiis/ DIIS methodcg-diis/ CG-DIIS method
- ncg (integer, Default=4)
- Available for 'dft' and 'dft_md' options of
theory.Number of interation of Conjugate-Gradient method for each scf-cycle.
- ncg_init (integer, Default=4)
- Available for 'dft' and 'dft_md' options of
theory.Number of interation of Conjugate-Gradient method for the first SCF step.
- method_mixing (character, Default='broyden')
- Available for 'dft' and 'dft_md' options of
theory.Methods for density/potential mixing for scf cycle.Optionssimple/ Simple mixing methodbroyden/ modified-Broyden methodpulay/ Pulay method
- mixrate (real(8), Default=0.5d0)
- Available for
method_mixing='simple'in 'dft' and 'dft_md' options oftheory.Mixing ratio for simple mixing.
- nmemory_mb (integer, Default=8)
- Available for
method_mixing='broyden'in 'dft' and 'dft_md' options oftheory.Number of previous densities to be stored in SCF iteration cycle for the modified-Broyden method. This must be less than 21.
- alpha_mb (real(8), Default=0.75d0)
- Available for
method_mixing='broyden'in 'dft' and 'dft_md' options oftheory.Parameter of the modified-Broyden method.
- nmemory_p (integer, Default=4)
- Available for
method_mixing='pulay'in 'dft' and 'dft_md' options oftheory.Number of previous densities to be stored in SCF iteration cycle for the Pulay method.
- beta_p (real(8), Default=0.75d0)
- Available for
method_mixing='pulay'in 'dft' and 'dft_md' options oftheory.Parameter of the mixing rate for the Pulay method.
- yn_auto_mixing (character, Default='n')
- Available for 'dft' and 'dft_md' options of
theory.The option to change the mixing-rate automatically (i.e. automatic adjustments ofmixrate/alpha_mb/beta_p)Options'y'/ enable'n'/ disable
- update_mixing_ratio (real(8), Default=3.0d0)
- Available for
yn_auto_mixing='y'in 'dft' and 'dft_md' options oftheory.Threshold for the change of the mixing-rate inyn_auto_mixing='y'option. The mixing-rate is reduced to half when the ratio of the density differences between the current and previous iteration steps is larger thanupdate_mixing_ratio.
- yn_subspace_diagonalization (character, Default='y')
- Available for 'dft' and 'dft_md' options of
theory.Option of subspace diagonalization during SCF cycle.Options'y'/ enable'n'/ disable
- convergence (character, Default='rho_dne')
- Available for 'dft' and 'dft_md' options of
theory.Quantity that is used for convergence check in SCF calculation.Options'rho_dne'/ Convergence is checked by sum_ix|rho(ix,iter)-rho(ix,iter-1)|dx/N, where iter is iteration number of SCF calculation and N is&system/nelec, the number of the valence electrons.'norm_rho'/ Convergence is checked by the square of the norm of difference of density, ||rho_iter(ix)-rho_iter-1(ix)||2=sum_ix|rho(ix,iter)-rho(ix,iter-1)|2.'norm_rho_dng'/ Convergence is checked by ||rho_iter(ix)-rho_iter-1(ix)||2/(number of grids). "dng" means "devided by number of grids".'norm_pot'/ Convergence is checked by ||Vlocal_iter(ix)-Vlocal_iter-1(ix)||2, where Vlocal is Vh + Vxc + Vps_local.'pot_dng'/ Convergence is checked by ||Vlocal_iter(ix)-Vlocal_iter-1(ix)||2/(number of grids).
- threshold (real(8), Default=1d-17 [a.u.] (for
convergence='rho_dne') and -1 (for other options ofconvergence)) - Available for 'dft' and 'dft_md' options of
theory.Threshold for convergence that is specified byconvergencekeyword.Unit conversions are: 1 a.u.= 45.54 A-6forconvergence='norm_rho'and'norm_rho_dng', 1 a.u.= 33.72x104A-6eV2forconvergence='norm_pot'and'norm_pot_dng'
- threshold (real(8), Default=1d-17 [a.u.] (for
- nscf_init_redistribution (integer, Default=10)
- Available for 'dft' and 'dft_md' options of
theory.The number of initial iterations for redistribution of the occupation number in finite temperature calculation.
- nscf_init_no_diagonal (integer, Default=10)
- Available for
&scf/yn_subspace_diagonalization='y'with 'dft' option oftheory.The number of initial iterations for which subspace diagonalization is not done.
- nscf_init_mix_zero (Integer, Default=-1)
- Available for 'dft' option of
theory.The densities is not mixed (i.e. fixed) during the given number of the SCF iteration cycle, that is, wavefunctions are optimized without updating the density.
- conv_gap_mix_zero (real(8), Default=99999d0)
- Available for positive number of
nscf_init_mix_zerowith 'dft' option oftheory.The condition to quite the fixed density iteration forced by
step_initial_mix_zerooption. The density is allowed to start mixing after the band-gap energy exceeds this given gap threshold for consecutive five SCF iteration steps,
&emfield¶
- trans_longi (character, Default='tr')
- Available for
yn_periodic='y'with 'maxwell' and TDDFT based options oftheory.Boundary condition for fields on macro-scale in solid-state calculations.Options'tr'/ Transverse'lo'/ longitudinal'2d'/ 2D maxwell-TDDFT method (for more details, seefilm_thicknessof &maxwell)
- ae_shape1/ae_shape2 (character, Default='none')
- Available for 'maxwell' and TDDFT based options of
theory.Envelope shape of the first/second pulse.Options'impulse'/ Impulsive fields.'Acos2'/ Envelope of cos2for a vector potential.'Acos3'/ Envelope of cos3for a vector potential.'Acos4'/ Envelope of cos4for a vector potential.'Acos6'/ Envelope of cos6for a vector potential.'Acos8'/ Envelope of cos8for a vector potential.'Ecos2'/ Envelope of cos2for a electric field.'Asin2cos'[Trial] / Envelope of sin2with cosine type oscillation for a vector potential.'Asin2_cw'[Trial] / Envelope of sin2at beginning and continuous wave after that for a vector potential (for 'ae_shape1' only).'input'[Trial] / read-in user-defined field is used given byfile_input1option (for 'ae_shape1' only).'none'/ no incident field is appliedFor
yn_periodic='n','impulse','Acos2', and'Ecos2'can be chosen.
- file_input1 (character, Default='')
- Available for
theory='tddft_pulse'withae_shape1='input'.The input file name for user-defined incident field (vector potential) when
ae_shape1='input'is used. The file must be numerical table (separated by blank) having more than four columns; the first column is time and second to fourth columns are Ax/c, Ay/c, Az/c, repsectively. All the quantities are written in units specified byunit_system, and '#' and '!' are available for a comment line. Besides, the linear interpolation is performed when the time step is differ from the calculation.
- e_impulse (real(8), Default=1d-2 a.u.)
- Available for 'maxwell' and TDDFT based options of
theory.Momentum of impulsive perturbation. This valiable has the dimention of momentum, energy*time/length.
- E_amplitude1/E_amplitude2 (real(8), Default=0d0)
- Available for 'maxwell' and TDDFT based options of
theory.Maximum amplitude of electric fields for the first/second pulse.This valiable has the dimension of electric field, energy/(length*charge). This cannot be set with
&emfield/I_wcm2_1(I_wcm2_2) simultaneously.
- I_wcm2_1/I_wcm2_2 (real(8), Default=-1d0)
- Available for 'maxwell' and TDDFT based options of
theory.Peak intensity (W/cm2) of the first/second pulse.This valiable cannot be set with
&emfield/E_amplitude1(E_amplitude2) simultaneously.
- tw1/tw2 (real(8), Default=0d0)
- Available for 'maxwell' and TDDFT based options of
theory.Duration of the first/second pulse (edge-to-edge time length).
- omega1/omega2 (real(8), Default=0d0)
- Available for 'maxwell' and TDDFT based options of
theory.Mean photon energy (average frequency multiplied by the Planck constant) of the first/second pulse.
- epdir_re1(3)/epdir_re2(3) (real(8), Default=1d0, 0d0, 0d0)
- Available for 'maxwell' and TDDFT based options of
theory.Real part of polarization unit vector for the first/second pulse.
- epdir_im1(3)/epdir_im2(3) (real(8), Default=0d0)
- Available for 'maxwell' and TDDFT based options of
theory.Imaginary part of polarization unit vector for the first/second pulse.
- phi_cep1/phi_cep2 (real(8), Default=0d0/0d0)
- Available for 'maxwell' and TDDFT based options of
theory.Carrier emvelope phase of the first/second pulse.
- t1_t2 (real(8), Default=0d0)
- Available for 'maxwell' and TDDFT based options of
theory.Time-delay between the first and the second pulses.
- t1_start (real(8), Default=0d0)
- Available for 'maxwell' and TDDFT based options of
theory.Time-delay of the first pulse. (this is not available for multiscale option).
- num_dipole_source (integer, Default=0)
- Available for TDDFT based options of
theory.Number of radiation sources for exciting optical near fields as incident sources. Maximum number is
2.
- vec_dipole_source(3,num_dipole_source) (real(8), Default=0d0)
- Available for TDDFT based options of
theory.Dipole vectors of the radiation sources for exciting optical near fields as incident sources.
- cood_dipole_source(3,num_dipole_source) (real(8), Default=0d0)
- Available for TDDFT based options of
theory.Central coordinates of the dipole vectors for exciting optical near fields as incident sources.
- rad_dipole_diele (real(8), Default=2d0 a.u.)
- Available for TDDFT based options of
theory.Radii of dielectric spheres for exciting optical near fields as incident sources.
&singlescale¶
- method_singlescale (character, Default='3d')
- Available for
theory='single_scale_maxwell_tddft'.Type of single-scale Maxwell-TDDFT method.Options:'3d'/ 3-dimensional FDTD + TDDFT'1d'/ 1-dimensional FDTD (along the z axis) + TDDFT'1d_fourier'/'1d'with 3D Fourier component of the vector potential
- cutoff_G2_emfield (real(8), Default=-1d0)
- Available for
theory='single_scale_maxwell_tddft'.Cutoff energy of Fourier component of the vector potential when method_singlescale='1d_fourier'.
- yn_symmetrized_stencil (character, Default='n')[Trial]
- Available for
theory='single_scale_maxwell_tddft'.Flag for the symmetrized finite differences of the product of the vector potential and the orbital wavefunction (nabla A(r) psi(r)).This option improves hermiticity of the Hamiltonian but makes worse the computational cost.
- yn_put_wall_z_boundary (character, Default='n')[Trial]
- Available for DFT/TDDFT based options of
theory.Option to put potential wall on the boundary plane at z=0 and z=``&system/al(3)``. This is to prevent the electrons from crossing the z-boundary plane. In the single-scale + Maxwell method, the electron density on the z-boundary plane can make the norm conservation (of electrons) less accurate due to the discontinuity of the vectorpotential. The wall is given by the square of cosine function.Options'y'/ put the potential wall'n'/ no potential wall
- wall_height (real(8), Default=100.0 eV)
- Available for
yn_put_wall_z_boundary='y'.The height of the potential wall.
- wall_width (real(8), Default=5.0 angstrom)
- Available for
yn_put_wall_z_boundary='y'.The width of the potential wall defined by the length from the potential peak (z=0 and z=``&system/al(3)``) to the edge.
&multiscale¶
- fdtddim (character, Default='1d')[Trial]
- Available for
theory='multi_scale_maxwell_tddft'withyn_periodic='y'Dimension of macroscopic scale system (Maxwell(FDTD) calculation) in multi-scale Maxwell-TDDFT method.Options:'3d'/ 3-dimensional FDTD for macroscopic scale (currently not available)'1d'/ 1-dimensional FDTD (along the x axis) for macroscopic scale
- nx_m (integer, Default=1)
- Available for
theory='multi_scale_maxwell_tddft'withyn_periodic='y'Number of macroscopic grid points inside materials for x-direction.
- ny_m/nz_m (integer, Default=1)[Trial]
- Available for
theory='multi_scale_maxwell_tddft'withyn_periodic='y'Number of macroscopic grid points inside materials for (y/z)-direction.
- hx_m (real(8), Default=0d0)
- Available for
theory='multi_scale_maxwell_tddft'withyn_periodic='y'Spacing of macroscopic grid points inside materials for (x)-direction. Unit of length can be chosen by
&units/unit_length. Variablehx_mis deprecated, and will be moved to&units/dl_em(1)
- hy_m/hz_m (real(8), Default=0d0)[Trial]
- Available for
theory='multi_scale_maxwell_tddft'withyn_periodic='y'Spacing of macroscopic grid points inside materials for (y/z)-direction. Unit of length can be chosen by
&units/unit_length. Variablehy_mandhz_mare deprecated, and will be moved to&units/dl_em(2:3)
- nxvacl_m/nxvacr_m (integer, Default=1/0)
- Available for
theory='multi_scale_maxwell_tddft'withyn_periodic='y'Number of macroscopic grid points for vacumm region.
nxvacl_mandnxvacr_mgive the number for positive x-direction in front of material,
&maxwell¶
- al_em(3) (real(8), Default=0d0)
- Available for
theory='maxwell'.Size of simulation box in electromagnetic analysis. Unit of the length can be chosen by
&units/unit_system.
- dl_em(3) (real(8), Default=0d0)
- Available for
theory='maxwell'andtheory='multi_scale_maxwell_tddft'.Spacing of real-space grids in electromagnetic analysis. Unit of length can be chosen by
&units/unit_system.
- dt_em (real(8), Default=0)
- Available for
theory='maxwell'.Time step in electromagnetic analysis. Unit of time can be chosen by
&units/unit_system.
- nt_em (integer, Default=0)
- Available for
theory='maxwell'.Number of total time steps for real-time propagation in electromagnetic analysis.
- boundary_em(3,2) (character, Default='default')
- Available for
theory='maxwell'andtheory='multi_scale_maxwell_tddft'.Boundary condition in electromagnetic analysis. The first index(1-3 rows) corresponds to x, y, and z axes. The second index(1-2 columns) corresponds to bottom and top of the axes. If
&system/yn_periodic='n','default','abc', and'pec'can be chosen, where'default'automatically chooses'abc'. If&system/yn_periodic='y','default','abc', and'periodic'can be chosen, where'default'automatically chooses'periodic'.'abc'is absorbing boundary,'pec'is perfect electric conductor, and'periodic'is periodic boundary. Whentheory='maxwell', perfectly matched layer(PML) is used as'abc'.
- shape_file (character, Default='none')
- Available for
theory='maxwell'.Name of input shape file in electromagnetic analysis. The shape file can be generated by using
FDTD_make_shapein SALMON utilities (https://salmon-tddft.jp/utilities.html).
- media_num (integer, Default=0)
- Available for
theory='maxwell'.Number of media in electromagnetic analysis.
- media_type(:) (character, Default='vacuum')
- Available for
theory='maxwell'.media_type(n)spesifies type of n-th media in electromagnetic analysis.'vacuum','constant media','pec', and'lorentz-drude'can be chosen. If'lorentz-drude'is chosen, linear response calculation can be done by&emfield/ae_shape1 or ae_shape2='impulse'.
- epsilon_em(:) (real(8), Default=1d0)
- Available for
theory='maxwell'.epsilon_em(n)spesifies relative permittivity of n-th media in electromagnetic analysis.
- mu_em(:) (real(8), Default=1d0)
- Available for
theory='maxwell'.mu_em(n)spesifies relative permeability of n-th media in electromagnetic analysis.
- sigma_em(:) (real(8), Default=0d0)
- Available for
theory='maxwell'.sigma_em(n)spesifies conductivity of n-th media in electromagnetic analysis.
- pole_num_ld(:) (integer, Default=1)
- Available for
theory='maxwell'.pole_num_ld(n)spesifies number of poles of n-th media for the case oftype_media(n)='lorentz-drude'in electromagnetic analysis.
- omega_p_ld(:) (real(8), Default=0d0)
- Available for
theory='maxwell'.omega_p_ld(n)spesifies plasma frequency of n-th media for the case oftype_media(n)='lorentz-drude'in electromagnetic analysis.
- f_ld(:,:) (real(8), Default=0d0)
- Available for
theory='maxwell'.f_ld(n,m)spesifies m-th oscillator strength of n-th media for the case oftype_media='lorentz-drude'in electromagnetic analysis. The first index is media ID id whose maximum value is determined bymedia_num. The second index is pole ID whose maximum value is determined bypole_num_ld(n).
- gamma_ld(:,:) (real(8), Default=0d0)
- Available for
theory='maxwell'.gamma_ld(n,m)spesifies m-th collision frequency of n-th media for the case oftype_media(n)='lorentz-drude'in electromagnetic analysis. The first index is media ID id whose maximum value is determined bymedia_num. The second index is pole ID whose maximum value is determined bypole_num_ld(n).
- omega_ld(:,:) (real(8), Default=0d0)
- Available for
theory='maxwell'.omega_ld(n,m)spesifies m-th oscillator frequency of n-th media for the case oftype_media(n)='lorentz-drude'in electromagnetic analysis. The first index is media ID id whose maximum value is determined bymedia_num. The second index is pole ID whose maximum value is determined bypole_num_ld(n).
- wave_input (character, Default='none')
- Available for
theory='maxwell'.If
'source', the incident pulse in electromagnetic analysis is generated by the incident current source.
- ek_dir1(3)/ek_dir2(3) (real(8), Default=0d0)
- Available for
theory='maxwell'.Propagation direction of the first/second pulse.
- source_loc1(3)/source_loc2(3) (real(8), Default=0d0)
- Available for
theory='maxwell'.Location of the incident current source of the first/second pulse. Note that the coordinate system ranges from
-al_em/2toal_em/2for&system/yn_periodic='n'while ranges from0toal_emfor&system/yn_periodic='y'.
- obs_num_em (integer, Default=0)
- Available for
theory='maxwell'.Number of observation point in electromagnetic analysis. From the obtained results, figure and animation files can be generated by using SALMON utilities (https://salmon-tddft.jp/utilities.html).
- obs_samp_em (integer, Default=1)
- Available for
theory='maxwell'.Sampling time-step of the observation in electromagnetic analysis.
- obs_loc_em(:,3) (real(8), Default=0d0)
- Available for
theory='maxwell'.obs_loc_em(n,1:3)=x,y,zspesifies location of n-th observation point in electromagnetic analysis. Note that the coordinate system ranges from-al_em/2toal_em/2for&system/yn_periodic='n'while ranges from0toal_emfor&system/yn_periodic='y'.
- yn_obs_plane_em(:) (character, Default='n')
- Available for
theory='maxwell'.Enable(
'y')/disable('n').yn_obs_plane_em(n)spesifies output of the electrmagnetic fields on the planes (xy, yz, and xz planes) for n-th observation point. This option must be'y'for generating animation files by usingFDTD_make_figaniin SALMON utilities (https://salmon-tddft.jp/utilities.html).
- yn_obs_plane_integral_em(:) (character, Default='n')
- Available for
theory='maxwell'.Enable(
'y')/disable('n').yn_obs_plane_integral_em(n)spesifies output of the spatial integration of electrmagnetic fields on the planes (xy, yz, and xz planes) for n-th observation point.
- yn_wf_em (character, Default='y')
- Available for
theory='maxwell'.Enable(
'y')/disable('n'). Applying a window function for linear response calculation when&calculation/theory=maxwell.
- film_thickness (real(8), Default=0d0)
- Available for TDDFT based options of
theorywithtrans_longi='2d'.Thickness of the film for the 2D maxwell-TDDFT method. The relative permittivity of the transparent media on both sides of the film can be specified by
epsilon_em(1)andepsilon_em(2), respectively.
- media_id_pml(3,2) (integer, Default=0)
- Available for
theory='maxwell'.Media ID used in PML. The first index(1-3 rows) corresponds to x, y, and z axes. The second index(1-2 columns) corresponds to bottom and top of the axes.
- media_id_source1/media_id_source2 (integer, Default=0)
- Available for
theory='maxwell'.Media ID used in incident current source1/source2 to generate the first/second pulse.
&analysis¶
- projection_option / out_projection_step (character/integer, Default='no'/100)
- Available for TDDFT based options of
theory.Methods of projection to analyze the excited states (e.g. the number of excited electrons.)Options'no'/ no projection.'gs'/ projection to eigenstates of ground-state Hamiltonian.'rt'/ projection to eigenstates of instantaneous Hamiltonian. [currently not available]This is printed evertyout_projection_stepstep during time-propagation.
- nenergy (integer, Default=1000)
Number of energy grid points for frequency-domain analysis. This parameter is used, for examples,
theory='tddft_response'andtheory='maxwell'.
- de (real(8), Default=0.01d0 eV)
Energy grid size for frequency-domain analysis. This parameter is used, for examples,
theory='tddft_response'andtheory='maxwell'.
- out_rt_energy_step (integer, Default=10)
- Available for the TDDFT based option of
theory.Total energy is calculated and printed every
out_rt_energy_steptime steps.
- yn_out_psi (character, Default='n')
- Available for
theory='dft'.Option for output of wavefunctionsOptions'y'/ enable.'n'/ disable.The format is specified by &analysis/
format_voxel_data.
- yn_out_dos (character, Default='n')
- Available for
theory='dft'.Option for output of density of stateOptions'y'/ enable.'n'/ disable.
- yn_out_pdos (character, Default='n')
- Available for
theory='dft'.Option for output of projected density of stateOptions'y'/ enable.'n'/ disable.
- yn_out_dos_set_fe_origin (character, Default='n')
- Available for
yn_out_dos='y'andyn_out_pdos='y'.Options to set the Fermi energy to zero'y'/ enable'n'/ disable.This option is not used if
&system/nstateis equal to&system/nelec/2.
- out_dos_start / out_dos_end (real(8), Default=-1d10 / 1d10 eV)
- Available for
yn_out_dos='y'andyn_out_pdos='y'.Lower/Upper bound (energy) of the density of state spectra. If this value is lower/higher than a specific value near the lowest/highest energy level, this parameter is re-set to the value.
- out_dos_nenergy (integer, Default=601)
- Available for
yn_out_dos='y'andyn_out_pdos='y'.Number of energy points sampled in the density of state spectra.
- out_dos_function (character, Default='gaussian')
- Available for
yn_out_dos='y'andyn_out_pdos='y'.Choise of smearing method for the density of state spectra.Options:gaussian/ Gaussian function is used.lorentzian/ Lorentzian function is used.
- out_dos_width (real(8), Default=0.1d0 eV)
- Available for
yn_out_dos='y'andyn_out_pdos='y'.Smearing width used in the density of state spectra.
- yn_out_dns (character, Default='n')
- Available for
theory='dft'.Option to print the spatial electron density distribution in the ground state.'y'/ enable'n'/ disable.
- yn_out_dns_rt/out_dns_rt_step (Character/Integer, Default='n'/50)
- Available for
theory='dft_md','tddft_pulse'.Options to print the spatial electron density distribution evertyout_dns_rt_stepstep during time-propagation.'y'/ enable'n'/ disable.
- yn_out_dns_ac_je/out_dns_ac_je_step (Character/Integer, Default='n'/50)
- Available for
theory='single_scale_maxwell_tddft'.Options to print the electron density, vector potential, electronic current, and ionic coordinates everyoutdns_dns_ac_je_steptime steps.'y'/ enable'n'/ disable.The data written in binary format are divided to files corresponding to the space-grid parallelization number.
- yn_out_dns_trans/out_dns_trans_energy (Character/Real(8), Default='n'/1.55d0eV)[currently not available]
- Available for
theory='tddft_pulse'.Option to calculate transition in different density from the ground state at specified frequency omega(given byout_dns_trans_energy) by drho(r,omega)=FT(rho(r,t)-rho_gs(r))/T.'y'/ enable'n'/ disable.(currently not available)
- yn_out_elf (character, Default='n')
- Available for
theory='dft'.Option to print the electron localization function.'y'/ enable'n'/ disable.
- yn_out_elf_rt/out_elf_rt_step (Character/Integer,Default='n'/50)
- Available for
theory='dft_md', 'tddft_pulse'.Option to print the electron localization function during the time-propagation everyout_elf_rt_steptime steps.'y'/ enable'n'/ disable.
- yn_out_estatic_rt/out_estatic_rt_step (Character/Integer, Default='n'/50)
- Available for
theory='tddft_pulse'.Option to print the static electric field during the time-propagation everyout_estatic_rt_steptime steps.'y'/ enable'n'/ disable.
- yn_out_rvf_rt/out_rvf_rt_step (Character/Integer, Default='n'/10)
- Available for TDDFT based options and 'dft_md' option of
theory.Option to print the coordinates[A], velocities[au], forces[au] on atoms during time-propagation inSYSname_trj.xyz everyout_rvf_rt_steptime steps.'y'/ enable'n'/ disable.If
yn_md='y', the printing option is automatically turned on.
- yn_out_tm (character, Default='n')[Trial]
- Available for
yn_periodic='y'withtheory='dft'.Option to calculate and print the transition moments between occupied and virtual orbitals toSYSname_tm.data after the ground state calculation.'y'/ enable'n'/ disable.
- out_ms_step (integer, Default=100)
- Available for
theory='multi_scale_maxwell_tddft'.Option to print some information everyout_ms_steptime step in the Maxwell + TDDFT multi-scale calculation.
- format_voxel_data (character, Default='cube')
- Available for
yn_out_psi='y',yn_out_dns(_rt)='y',yn_out_dns_ac_je='y',yn_out_elf(_rt)='y',yn_out_estatic_rt='y'.Option of the file format for three-dimensional volumetric data.'avs'/ AVS format'cube'/ cube format'vtk'/ vtk format
- nsplit_voxel_data (integer, Default=1)
- Available for
format_voxel_data='avs'.Number of separated files for three dimensional data.
- yn_out_perflog (character(1), Default='y')
- Available for all
theoryOption to print the performance log of routines and modules.
- format_perflog (character(6), Default='stdout')
- Available for
yn_out_perflog = 'y'The output format of performance log.'stdout'/ standard output unit'text'/ save to text file'csv'/ save to csv format file
&poisson¶
- layout_multipole (character, Default=3)
- Available for
yn_periodic='n'with DFT and TDDFT based options oftheory.A variable to determine how to put multipoles in the Hartree potential calculation.
Options:1/ A single pole is put at the center.2/ Multipoles are put at the center of atoms.3/ Multipoles are put at the center of mass of electrons in prepared cuboids.
- num_multipole_xyz(3) (integer, Default=0)
- Available for
yn_periodic='n'with DFT and TDDFT based options oftheory.Number of multipoles. When default is set, number of multipoles is calculated automatically.
- lmax_multipole (integer, Default=4)[Trial]
- Available for
yn_periodic='n'with DFT and TDDFT based options oftheory.A maximum angular momentum for multipole expansion in the Hartree-cg calculation.
- threshold_cg (real(8), Default=1d-15 a.u.(= 1.10d-13 A^3eV^2))
- Available for
yn_periodic='n'with DFT and TDDFT based options oftheory.A convergence value for the Hartree-cg calculation. The convergence is checked by ||tVh(i)-tVh(i-1)||^2/(number of grids).
&ewald¶
- newald (integer, Default=4)
- Available for
yn_periodic='y'with DFT/TDDFT based options oftheory.Parameter for Ewald method for ion-ion Coulombic interaction. Short-range part of Ewald sum is calculated withinnewaldth nearlist neighbor cells.
- aewald (real(8), Default=0.5d0)
- Available for
yn_periodic='y'with DFT/TDDFT based options oftheory.Square of range separation parameter for Ewald method in atomic unit.
- cutoff_r (real(8), Default=-1d0)
- Available for
yn_periodic='y'with DFT/TDDFT based options oftheory.Cut-off length in real-space. This is automatically chosen in default (negative number)
- cutoff_r_buff (real(8), Default=2d0 a.u.)
- Available for
yn_periodic='y'withyn_md='y'ortheory='dft_md'.Buffer length in radius for book-keeping for real-space interaction.
- cutoff_g (real(8), Default=-1d0)
- Available for
yn_periodic='y'with DFT/TDDFT based options oftheory.Cut-off in G-space in the Ewald method. No cut-off in default.
&opt[Trial]¶
- nopt (integer, Default=100)
- Available for
yn_opt='y'withtheory='dft'.The maximum step number of geometry optimization.
- convrg_opt_fmax (real(8), Default=1d-3 [a.u.])
- Available for
yn_opt='y'withtheory='dft'.Convergence threshold of geometry optimization in maximum force on atom.
- max_step_len_adjust (real(8), Default=-1d0)
- Available for
yn_opt='y'withtheory='dft'.Set maximum optimization step length (if positive number is given)
&md[Trial]¶
- ensemble (character, Default='NVE')
- Available for
yn_md='y'ortheory='dft_md'.Ensemble in MD option:Options:NVE/ NVE ensemble (constant energy and volume system)NVT/ NVT ensemble (constant temperature and volume system)
- thermostat (character, Default='nose-hoover')
- Available for
yn_md='y'ortheory='dft_md'.Thermostat in "NVT" option:Options:nose-hoover/ Nose-Hoover thermostat.
- step_velocity_scaling (integer, Default=-1)
- Available for
yn_md='y'ortheory='dft_md'.Time step interval for velocity-scaling. Velocity-scaling is applied if this is set to positive.
- step_update_ps (Integer, Default=10)
- Available for
yn_md='y'ortheory='dft_md'.Time step interval for updating pseudopotential (Larger number makes calculation time reduce but gets inaccurate).
- temperature0_ion_k (real(8), Default=298.15d0 [K])
- Available for
yn_md='y'ortheory='dft_md'.Setting ionic temperature [K] for NVT ensemble, velocity scaling and generating initial velocities.
- yn_set_ini_velocity (character, Default='n')
- Available for
yn_md='y'ortheory='dft_md'.Option to generate initial velocities.Options:y/ Generate initial velocity with Maxwell-Bortzman distribution.n/ disable.
- file_ini_velocity (character, Default='none')[Trial]
- Available for
yn_md='y'ortheory='dft_md'.File name for reading initial velocities. This is read if the file name is given, then, the priority is higher than use ofset_ini_velocityand restart data of velocities. The format is simply vx(iatom) vy(iatom) vz(iatom) in each line. The order of atoms must be the same as the given coordinates in the main input file. In case of using nose-hoover thermostat, a thermostat variable should be put at the last line (all atomic unit).
- thermostat_tau (real(8), Default=41.34d0 a.u. or 1d0 fs)
- Available for
yn_md='y'ortheory='dft_md'.Parameter in Nose-Hoover method: controlling time constant for temperature.
- yn_stop_system_momt (character, Default='n')
- Available for
yn_md='y'ortheory='dft_md'.Center of mass is fixed every time step.Options:y/ enable.n/ disable.
&jellium¶
- yn_jm (character, Default='n')
- Available for the DFT/TDDFT based options of
theory.Jellium model option:Options:y/ enable.n/ disable.When
yn_jm='y',&functional/xcmust be'pz'.
- yn_charge_neutral_jm (character, Default='y')
- Available for
yn_jm='y'with the DFT/TDDFT based options oftheory.Option to satisfy charge neutrality :Options:y/ enable.rs_bohr_jmis automatically modified so as to satisfy charge neutrality.n/ disable.rs_bohr_jmis not automatically modified but the calculation involves small charge neutrality error.
- yn_output_dns_jm (character, Default='y')
- Available for
yn_jm='y'with the DFT/TDDFT based options oftheory.Option to output positive background charge density:Options:y/ enable.n/ disable.
- shape_file_jm (character, Default='none')
- Available for
yn_jm='y'with the DFT/TDDFT based options oftheory.Name of input shape file to generate positive background charge density used in jellium model. The shape file can be generated by using
FDTD_make_shapein SALMON utilities (https://salmon-tddft.jp/utilities.html). Whenshape_file_jm='none', the shape of the positive background charge density is specified bysphere_nelec_jmandsphere_loc_jmwhich generate spherical shapes.
- num_jm (integer, Default=0)
- Available for
yn_jm='y'with the DFT/TDDFT based options oftheory.When
shape_file_jmis not 'none',num_jmspecifies number of media used in jellium model. Whenshape_file_jm='none',num_jmspecifies number of spherical shapes.
- rs_bohr_jm(:) (real(8), Default=0do)
- Available for
yn_jm='y'with the DFT/TDDFT based options oftheory.When
shape_file_jmis not 'none',rs_bohr_jm(n)spesifies the Wigner-Seitz radius for n-th media. Whenshape_file_jm='none',rs_bohr_jm(n)spesifies the Wigner-Seitz radius for n-th sphere.
- sphere_nelec_jm(:) (integer, Default=0)
- Available for
yn_jm='y'andshape_file_jm='none'with the DFT/TDDFT based options oftheory.sphere_nelec_jm(n)spesifies electron number for n-th sphere.
- sphere_loc_jm(:,3) (real(8), Default=0d0)
- Available for
yn_jm='y'andshape_file_jm='none'with the DFT/TDDFT based options oftheory.sphere_loc_jm(n,1:3)=x,y,zspesifies location of center of mass for n-th sphere. Note that the coordinate system ranges from-al/2toal/2for&system/yn_periodic='n'while ranges from0toalfor&system/yn_periodic='y'.
&code¶
- yn_want_stencil_hand_vectorization (character, Default='y')
- This option requests hand-vectorized optimization code of stencil in the hamiltonian calculation.SALMON checks the calculation can be used the hand-vectorized code.If failing it, SALMON will uses the typical implementation.
- yn_want_communication_overlapping (character, Default='n')
- Available for
theory='tddft*' or '*maxwell_tddft'This option requests computation/communication overlap algorithm to improve the performance of stencil in the hamiltonian calculation.SALMON checks the calculation can be used the overlap algorithm.If failing it, SALMON will uses the non-overlap algorithm.
- stencil_openmp_mode (character, Default='auto')
- This option selects a OpenMP parallelization mode of stencil in the hamiltonian calculation.
auto/ SALMON decides the parallelization target automatically.orbital/ OpenMP parallelization is applied to orbital (and k-point) loop.rgrid/ OpenMP parallelization is applied to real-space grid loop.
- current_openmp_mode (character, Default='auto')
- This option selects a OpenMP parallelization mode of the current calculation.
auto/ SALMON decides the parallelization target automatically.orbital/ OpenMP parallelization is applied to orbital (and k-point) loop.rgrid/ OpenMP parallelization is applied to real-space grid loop.
- force_openmp_mode (character, Default='auto')
- This option selects a OpenMP parallelization mode of the force calculation.
auto/ SALMON decides the parallelization target automatically.orbital/ OpenMP parallelization is applied to orbital (and k-point) loop.rgrid/ OpenMP parallelization is applied to real-space grid loop.